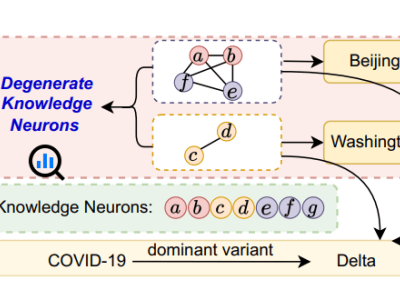
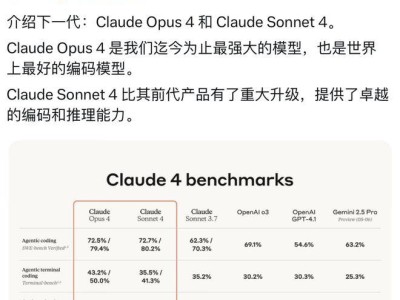
meta公司近期宣布了两项重大突破,为化学研究和应用开辟了新的道路。他们发布了OMol25,这是迄今为止规模最大的AI驱动化学开放数据集,并同步推出了UMA,一个通用的人工智能模型,专门用于预测分子和材料化学性质。
OMol25数据集包含了超过1亿次的分子计算数据,其规模远远超越了以往任何同类公开数据集。meta为此投入了超过60亿小时的计算资源,以确保数据的精确性和全面性。该数据集覆盖了广泛的分子类型,从小型有机化合物到生物分子,再到金属复合物和电解质,一应俱全。它还包含了分子的多种状态、空间排列以及化学反应的相关信息,提供了详尽的化学性质数据,如能量、力值、电荷分布和轨道等。目前,这一宝贵资源已在Hugging Face平台上向公众开放。
与OMol25数据集一同亮相的,是meta精心打造的UMA模型。这个模型基于OMol25以及其他数据集进行训练,其独特之处在于能够在原子层面快速预测化学性质,速度远超传统计算方法。与以往需要针对特定任务构建专门模型的方法不同,UMA具有高度的通用性,能够广泛应用于从分子模拟到材料和催化研究等多个领域。UMA采用了先进的图神经网络和“混合线性专家”架构,实现了计算速度和预测精度的完美平衡。在基准测试中,UMA的表现已经达到了以往只有精细调整的专用模型才能达到的水平。
meta强调,UMA模型的推出将极大地加速研究进程。以往需要数天才能完成的分子模拟和计算,现在借助UMA只需几秒钟即可完成。这意味着研究人员可以在实验室合成之前,快速筛选数千种潜在的新分子,从而高效评估其作为药物或电池材料的潜力。目前,UMA模型也已在Hugging Face平台上开放获取。
除了OMol25数据集和UMA模型外,meta还推出了一种名为“伴随采样”的全新AI分子模拟方法。这一方法打破了传统AI模型需要大量真实世界数据来生成新分子结构的限制。即使在缺乏真实样本的情况下,“伴随采样”也能学习并提出新的分子结构。该技术融合了随机控制理论和扩散过程的概念,meta团队认为扩散过程特别适合用于模拟分子。实验显示,“伴随采样”仅需少量计算就能快速探索多种分子结构变体,且生成的分子构象不仅能与传统软件的结果相匹配,在处理具有多个灵活组件的分子时甚至表现更为出色。相关的模型、代码和更多信息已在Hugging Face和GitHub平台上提供。
尽管取得了这些显著进展,meta也坦诚地指出了当前面临的挑战。例如,在聚合物、某些金属或复杂的质子化状态等化学领域,数据的覆盖尚不全面。AI模型在预测电荷、自旋和长程相互作用等性质方面仍有待提升。meta表示,他们将继续致力于解决这些问题,以推动化学研究和应用的进一步发展。